Homepage
For pipeline code visit HARMLINC GitHub Repo.
Overview
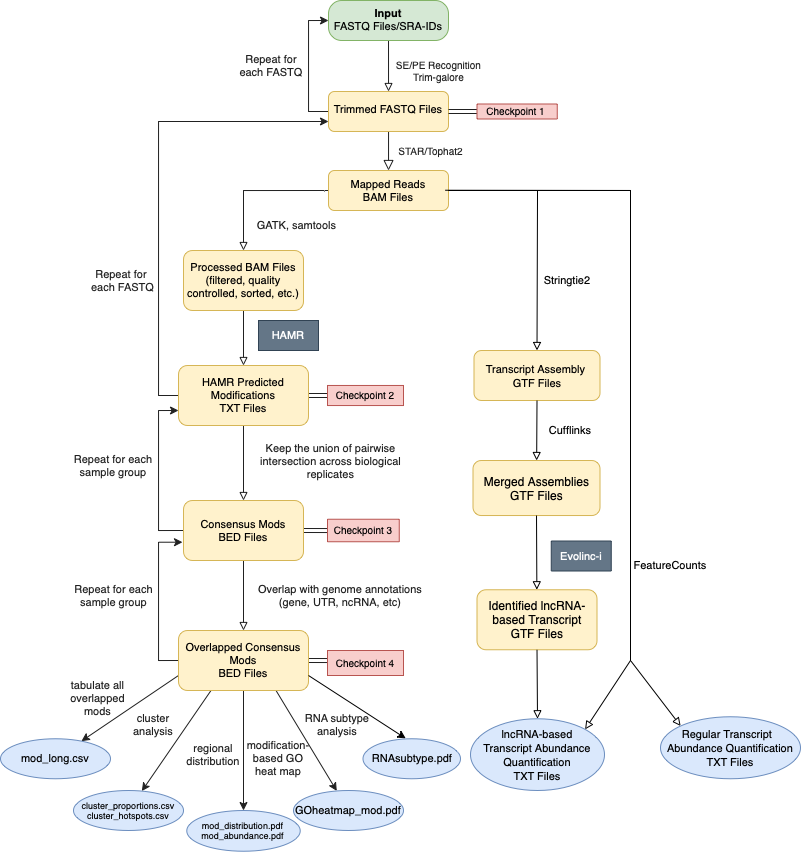
- HAMRLINC is a multipurpose toolbox that expedites the analysis pipeline of two algorithms: HAMR and Evolinc. The former was developed by Paul Ryvkin et al, and the latter by Andrew D.L. Nelson et al. HAMRLINC aims to make the original methods more accessible by automating the tedious pre-processing steps, and expand on their functionalities with its built-in post-processing steps, allowing users to perform RNA modification prediction with intuitive output formats, lincRNA identification and transcripts abundance quantifiction using the same RNA-Seq input data.
- HAMRLINC is high-throughput and performs RNA-modification analysis, long intergenic non-coding RNAs(lincRNA) annotation and transcripts abundance quantification at a bioproject scale. HAMRLINC performs constitutive trimming of acquired reads using Trim-Galore, and makes use of STAR (Tophat option available) as the default aligning tool; mapped reads are pre-processed using selected methods from GATK and samtools.
- HAMRLINC is optimized for partial parallel processing, and modularization. Specifying a larger thread count where hardware permits will greatly increase the efficiency of a run. If only partial functionality is needed (e.g. Only analyzing modified ribonucleotides), users can use flags to activate needed function modules as they desire. See below for more details .
HARMLINC Workflow